BrainVoyager v23.0
Anatomical Preprocessing Workflow
The anatomical preprocessing workflow can be used to perform brain extraction and spatial intensity inhomogeneity correction for each subject and session of a project.
Creating and Connecting the Workflow
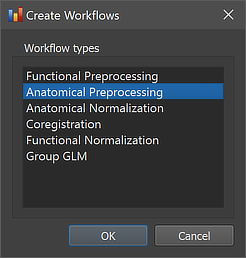
The anatomical preprocessing workflow can be created using the Create Workflows dialog that can be launched by using the Create button in the Workflows tab of the Data Analysis Manager window. In the appearing dialog, the Anatomical Preprocessing entry needs to be selected (see screenshot below). Note that one could add other workflows at the same time (e.g. "Anatomical Normalization") but here we describe the creation of each workflow in a separate step.
After clicking the OK button, the new workflow (the first for the current project) appears in the Workflows tab (see figure below). The ID column indicates that the unique identifier '1' has been assigned to this workflow; the column Type shows the generic name "Anatomical Preprocessing" for the assigned (and stored) type ID (2); the column Name shows the default name "anat preprocessing" that has been given to this workflow, which can be changed in the Workflow dialog.
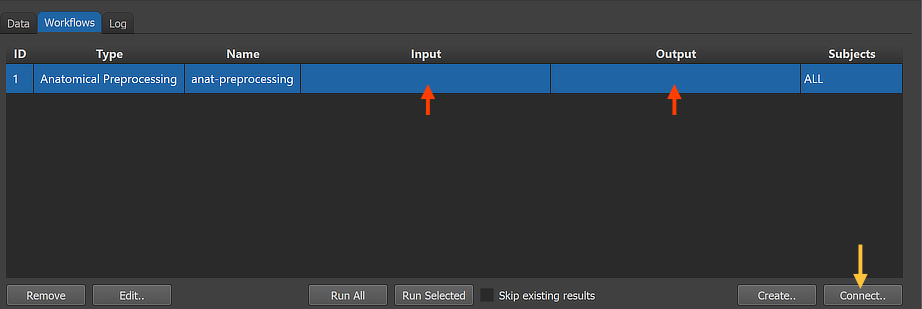
The red arrows in the figure above highlight that the instantiated preprocessing workflow is not yet ready to be executed since no data connections have been made. To connect the workflow to input data, the Connect Workflows dialog is used, which can be invoked by clicking the Connect button in the Workflows tab (see orange arrow above).
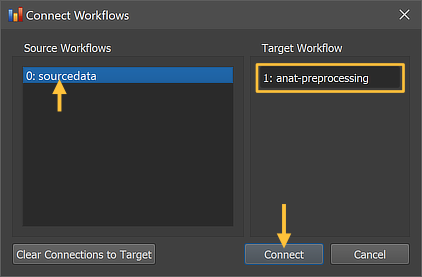
The Connect Workflows dialog shows in the Source Workflows list on the left side all available data sources.. A specific workflow is identified by its unique ID and its name ("[workflow-ID]: [workflow-name]"). On the right side the target workflow is displayed to which data can be connected which is the selected anatomical preprocessing workflow. Since no other workflow has been defined yet, only the original NIfTI source data ("0: sourcedata" entry) can be selected as the source data for the anatomical preprocessing (target) workflow. Note that no specific dataset (file name) needs to be specified since the program uses the predictable BIDS directory tree to find the appropriate anatomical source file (per session) in the respective "anat" folder of each subject.
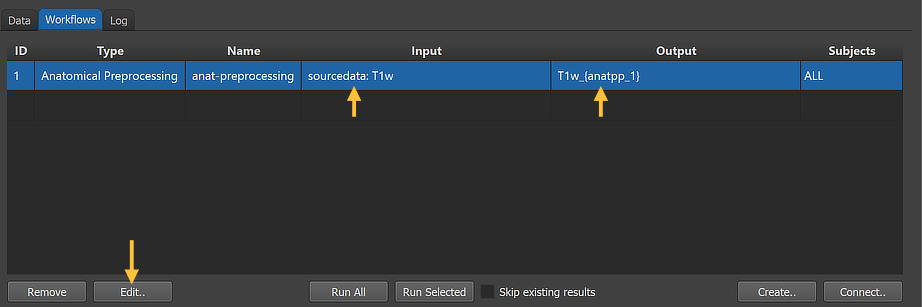
After clicking the Connect button in the Connect Workflows dialog, the established connection is made and the workflow entry is updated accordingly. The upper arrows in the figure above indicate that the anatomical preprocessing workflow will use the original NIfTI data in the sourcedata folder as input using a standard "T1w" (T1-weighted) file as input. The program has retrieved this name by checking the full NIfTI file name ('sub-01_ses-01_T1w.nii.gz') in the sourcedata folder of subject 1 (usually 'sub-01') as a proxy for all subjects and removed the subject and session IDs (as well as the file extension) since the workflow will process (if not restricted) the data from all subjects and across all sessions.
Inspecting and Editing Workflow Settings
In order to inspect and eventually change settings, such as parameters that control the processing, one can invoke the Workflow dialog by double-clicking the workflow entry or by clicking the Edit button (see arrow above).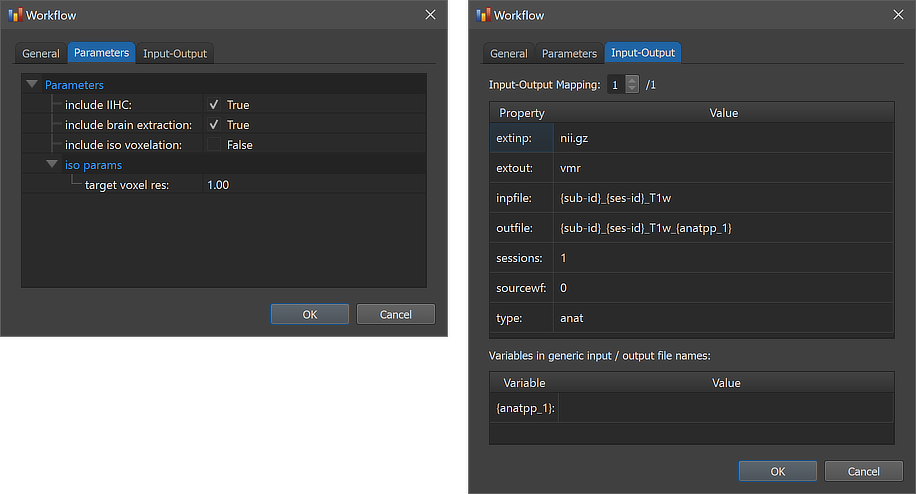
The figure above shows the contents of the Parameters tab (left) and he Input-Output tab (right) of the Workflow dialog. The Parameters tab shows the parameters specific for the selected workflow, which can be inspected as well as changed. The default preprocessing options set the include IIHC option to 'True', which implies that the standard intensity inhomogeneity correction (IIHC) step will be performed for each included dataset. The workflow will perform a "brain skull stripping" procedure since the include brain extraction option is set to 'True'. The include iso voxelation option is set to 'False' since the 3D anatomical datasets are expected to have a voxel size of 1mm x 1mm x 1mm. If this is not the case, the option should be set to 'True'. In that case the anatomical datasets will be rescaled to the resolution specified in the target voxel res field of the iso params settings, which has value '1.0' as default.
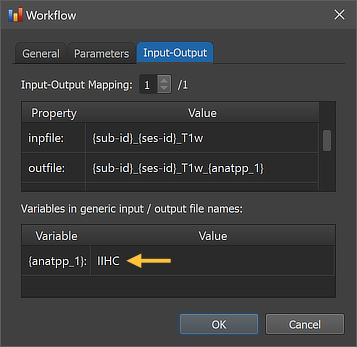
The right side of the figure above shows the Input-Output tab, which displays information related to the established data flow connection(s). Each connection (if more than 1) can be viewed by changing the Input-Output Mapping number. The details of the displayed fields are usually not important to know but it might sometimes be instructive to inspect these (read-only) fields. Probably most relevant are the inpfile and outfile fields, which show the processed T1-weighted anatomical file name with the subject and session substrings replaced by (loop) variables (enclosed by curly braces '{}'). The outfile entry has an extended file name that adds the substring "_{anatpp_1"; the processing variable "{anatpp_1}" is also visible in the Variables in generic input / output file names field (here loop variables, such as subject and session variables are not shown). When running the workflow, processing variables will be replaced with a concrete value (substring) that depends on the included processing steps and parameter settings. When inspecting the variables section in the dialog after running the workflow, the concrete values will be displayed. Note that variables (and their values if available) will be handed over to any subsequent workflow so that file names with additional substrings can be constructed before any workflow has been executed.
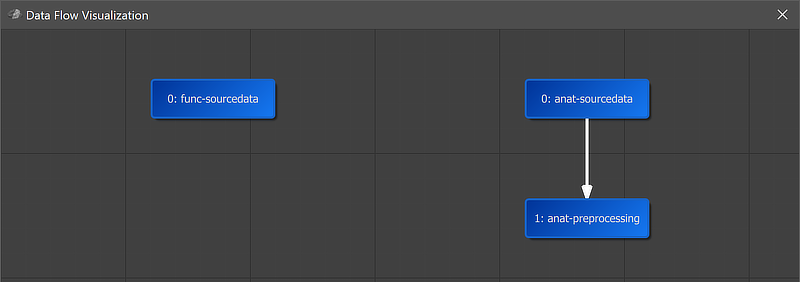
A global overview of the established connection is graphically displayed when clicking the Data Flow button in the main toolbar of the Data Analysis Manager (see screenshot above). In the appearing visualization the "1: anat-preprocessing" workflow is placed under the "0: anat-sourcedata" node and an arrow connects the anatomical source data with the preprocessing workflow.
Running the Workflow
After selecting the workflow in the Workflows tab (single-click on its row in the table), the prepared anatomical workflow can be executed using the Run Selected button (or the Run All) button). In case that the Skip existing results option has been turned on, the program will not process datasets in case that the expected output data are already available. This option is useful (saving time) in case that one adds more subjects to a project and wants to preprocess only the newly added ones. An alternative way to restrict the workflow to specific subjects is to select the desired subset of subjects in the Subjects field in the General tab of the Workflow dialog.
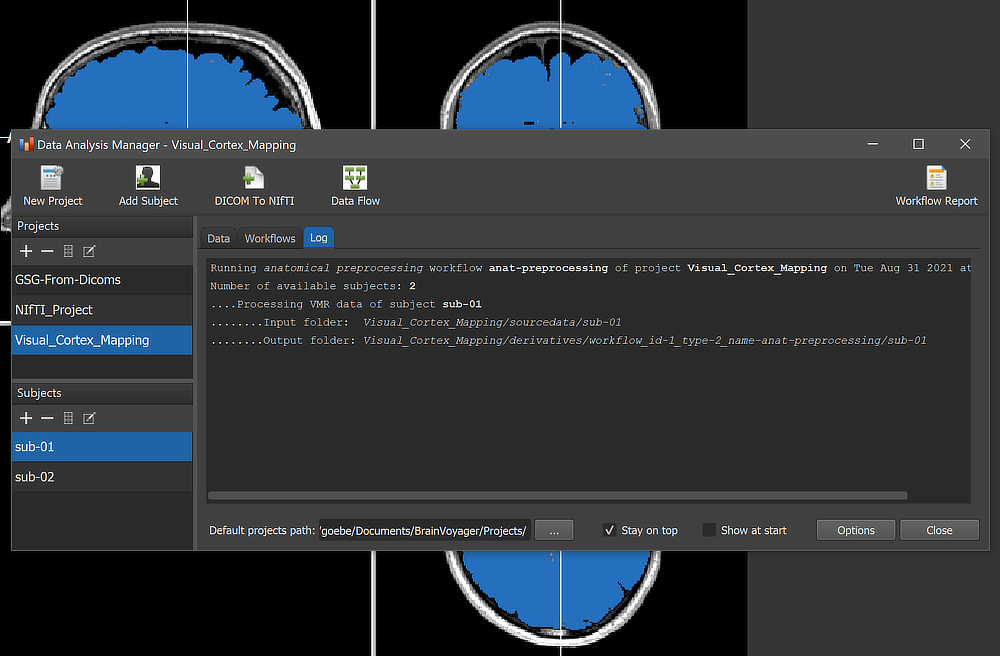
When running a workflow, the program displays the progress of data processing in the Log tab of the Data Analysis Manager window. The screenshot above captures a moment during anatomical preprocessing (brain extraction step for subject 'sub-01').
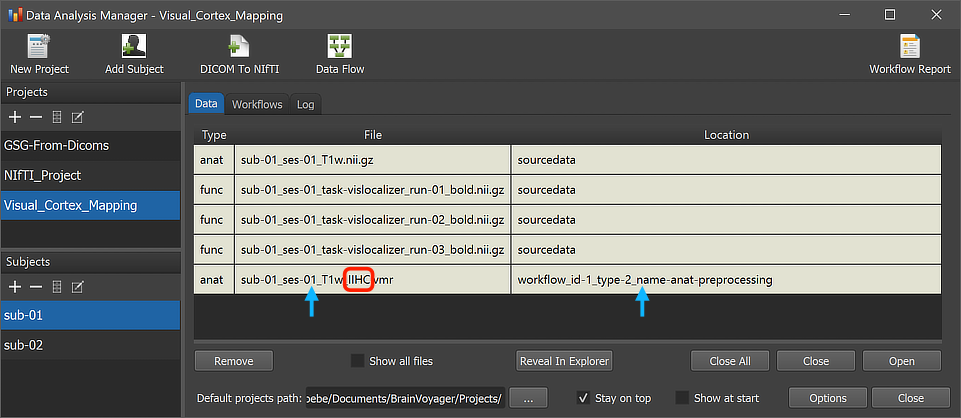
After the workflow has finished processing, the main produced data files can be inspected by switching to the Data tab of the Data Analysis Manager. The arrows in the screenshot above highlight the newly added entry for the selected subject but the same output will be available for all subjects (here only 2) in case that processing was successful. The red rectangle highlights that the produced output file name in the File column now has the additional substring "_IIHC" as compared to the input file name shown in the first row of the Subject Data table. The substring "IIHC" is also stored now as the value of the "{anatpp_1}" variable as shown in the Workflow dialog (see screenshot below).
The Location column of the Data table of the Data Analysis Manager indicates that the produced files are stored inside the created workflow directoy (under the derivatives folder of the project); the name of the workflow folder 'workflow_id-1_type-2_name-anat-preprocessing' contains the unique workflow ID ('1'), its type ('2') and its name ('anat-preprocessing'). Note that you can open the produced file by simply double-clicking its row in the Data table or by clicking the Open button below the table after selecting the respective table row. The location on disk of the generated file can be shown in the Finder (macOS) or Explorer (Windows) by clicking the Reveal in Finder or Reveal in Explorer button. The figure below shows the full generated directory tree of the workflow on disk.
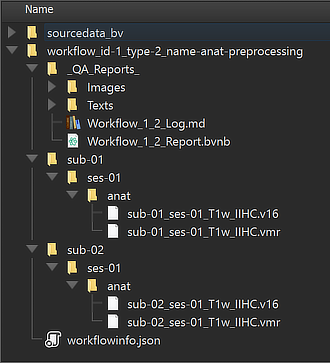
This view illustrates that a workflow on disk replicates the BIDS directory tree for the relevant data (here "anat" but no "func" or "dwi" folders). The file names in the "anat" folders reveal that the preprocessed output data are stored both in 8-bit (VMR) as well 16-bit (V16) format. Besides the BIDS sub-directories for each subject, the "_QA_Reports_" folder is also stored inside the workflow directory containing the full log (Markdown) text file and the quality report as a BrainVoyager notebook (see below), which will be shown automatically when the workflow has completed. The workflow report can also be shown at a later time by clicking the Workflow Report icon in the main toolbar of the Data Analysis Manager after selecting the respective entry in the Workflows table.
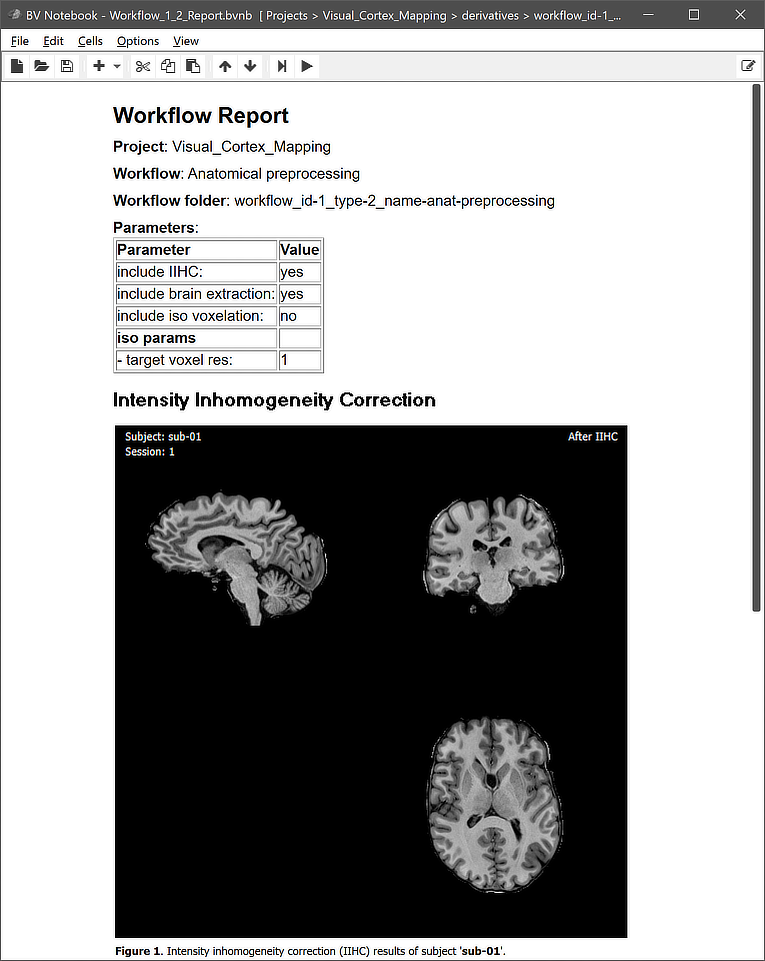
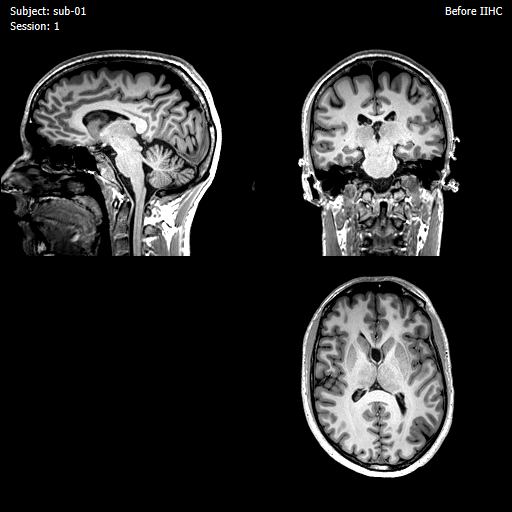
Note that the figures generated per subject are actually animations (not visible in screenshot above) showing the anatomical dataset before and after preprocessing. The animation embedded in the quality report for subject 'sub-01' is shown below.
Copyright © 2023 Rainer Goebel. All rights reserved.